TOGA_genes_per_scaffold
Juan M Vazquez (docmanny)
2022-12-05
Last updated: 2023-01-31
Checks: 7 0
Knit directory: R_workflowr/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221115) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version ab25732. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: R_workflowr/.RData
Ignored: R_workflowr/.Rhistory
Ignored: R_workflowr/.Rprofile
Ignored: R_workflowr/.Rproj.user/
Ignored: R_workflowr/_workflowr.yml
Ignored: analyses/annotation/output/
Ignored: analyses/cactus/data/
Ignored: analyses/pangenome/output/
Ignored: data/USGS_SpeciesRanges/
Ignored: data/genomes/
Ignored: data/gff/
Ignored: data/tree/
Untracked files:
Untracked: R_workflowr/.gitattributes
Untracked: R_workflowr/.gitignore
Untracked: R_workflowr/R_workflowr.Rproj
Untracked: R_workflowr/analysis/
Untracked: R_workflowr/ng_curves.html
Untracked: R_workflowr/renv.lock
Untracked: R_workflowr/renv/
Untracked: analyses/mMyoLuc1/
Untracked: analyses/makeHub/
Untracked: analyses/tmp-mMyoLuc1/
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Introduction
library(tidyverse)Warning in system("timedatectl", intern = TRUE): running command 'timedatectl'
had status 1── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──
✔ ggplot2 3.4.0 ✔ purrr 0.3.5
✔ tibble 3.1.8 ✔ dplyr 1.0.10
✔ tidyr 1.2.1 ✔ stringr 1.4.1
✔ readr 2.1.3 ✔ forcats 0.5.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(naturalsort)
library(ggpubr)toga.vel <- read_tsv("/mnt/c/users/manue/OneDrive/Desktop/query_annotation.bed", col_types = "cddcdcddcdcc",
col_names=c("chr", "s", "e", "name", "score", "strand", "thickStart", "thickEnd", "RBG", "nBlock", "blockStart", "blockLength" ))
toga.vel # A tibble: 186,006 × 12
chr s e name score strand thick…¹ thick…² RBG nBlock block…³
<chr> <dbl> <dbl> <chr> <dbl> <chr> <dbl> <dbl> <chr> <dbl> <chr>
1 SUPER_… 7.90e7 7.90e7 ENST… 1000 - 7.90e7 7.90e7 0,0,… 18 3,84,1…
2 SUPER_… 2.04e6 2.04e6 ENST… 1000 + 2.04e6 2.04e6 130,… 1 106,
3 SUPER_… 2.85e6 2.85e6 ENST… 1000 - 2.85e6 2.85e6 255,… 1 16,
4 SUPER_… 8.00e7 8.00e7 ENST… 1000 + 8.00e7 8.00e7 255,… 1 3,
5 SUPER_… 8.42e7 8.42e7 ENST… 1000 - 8.42e7 8.42e7 255,… 1 3,
6 SUPER_… 9.64e7 9.64e7 ENST… 1000 - 9.64e7 9.64e7 255,… 2 12,54,
7 SUPER_… 1.14e6 1.14e6 ENST… 1000 - 1.14e6 1.14e6 255,… 1 49,
8 SUPER_… 1.71e5 1.71e5 ENST… 1000 - 1.71e5 1.71e5 255,… 1 3,
9 SUPER_… 5.68e7 5.68e7 ENST… 1000 - 5.68e7 5.68e7 255,… 1 66,
10 SUPER_… 2.64e7 2.64e7 ENST… 1000 - 2.64e7 2.64e7 255,… 1 98,
# … with 185,996 more rows, 1 more variable: blockLength <chr>, and abbreviated
# variable names ¹thickStart, ²thickEnd, ³blockStartgenome.vel <- read_tsv("../../analyses/makeHub/data/genomes/mMyoVel1.genome", col_names = c("chr", "length"))Rows: 162 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): chr
dbl (1): length
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.genome.vel# A tibble: 162 × 2
chr length
<chr> <dbl>
1 SUPER__1 234804448
2 SUPER__2 216113371
3 SUPER__3 212800901
4 SUPER__4 118339641
5 SUPER__5 113646647
6 SUPER__6 110944935
7 SUPER__7 98831194
8 SUPER__8 93943467
9 SUPER__9 91941725
10 SUPER__10 86976347
# … with 152 more rowstoga.vel.summary <- toga.vel %>%
group_by(chr) %>%
summarize(n_genes = n_distinct(name)) %>%
left_join(genome.vel, by="chr") %>%
mutate(chr = factor(chr, levels=str_sort(chr, numeric = T) %>% unique))toga.vel.summary %>%
ggplot(
aes(x=length, y=n_genes, label=chr)
) +
geom_text(aes(color=(as.numeric(chr)<=23))) +
theme_pubclean() +
scale_x_log10() +
scale_y_log10() +
ggtitle("TOGA: mMyoVel1 (hg38)")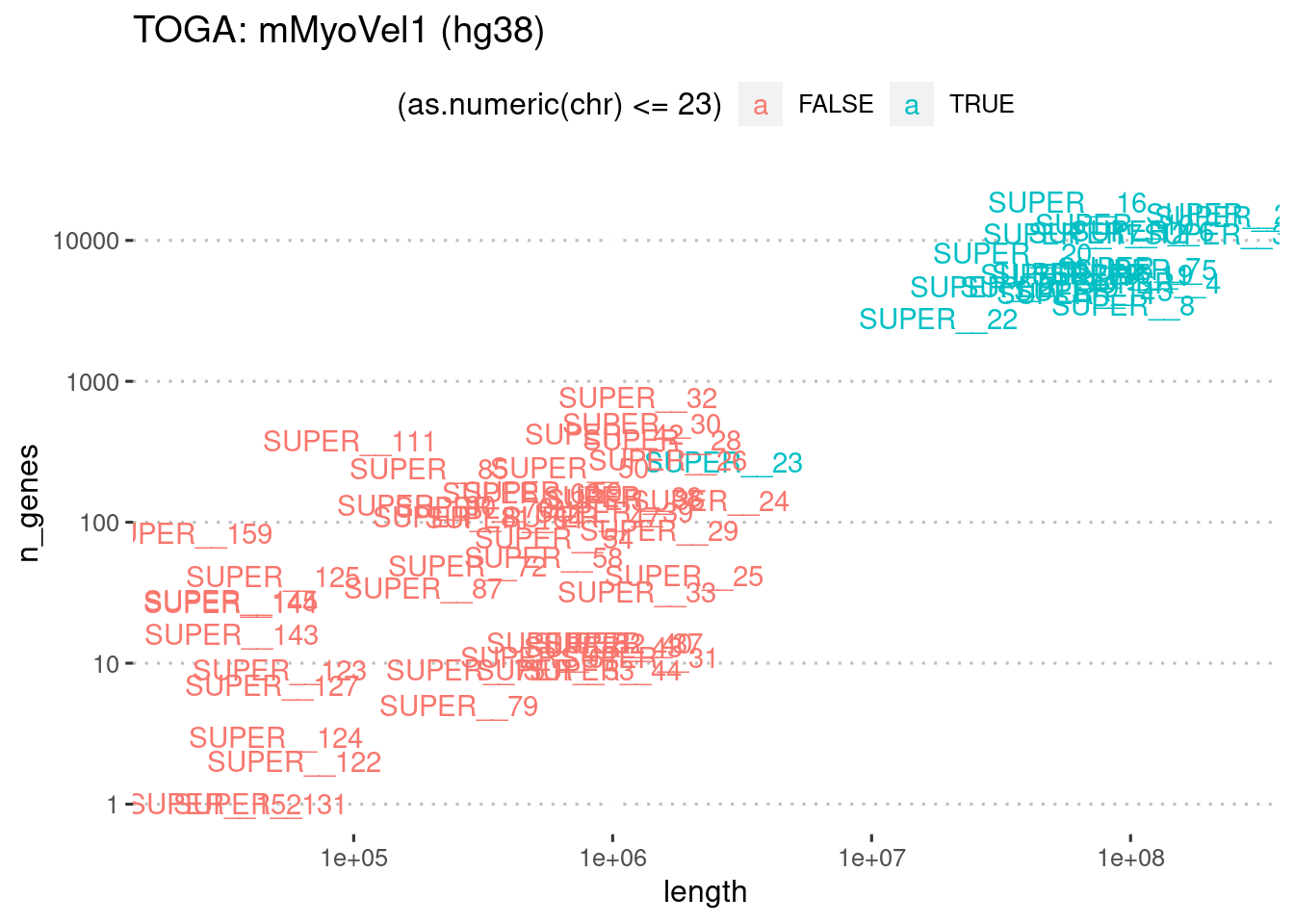
toga.luc <- read_tsv("/mnt/c/users/manue/OneDrive/Desktop/TOGA_mMyoLuc1_hg38.bed", col_types = "cddcdcddcdcc",
col_names=c("chr", "s", "e", "name", "score", "strand", "thickStart", "thickEnd", "RBG", "nBlock", "blockStart", "blockLength" ))
toga.luc # A tibble: 189,518 × 12
chr s e name score strand thick…¹ thick…² RBG nBlock block…³
<chr> <dbl> <dbl> <chr> <dbl> <chr> <dbl> <dbl> <chr> <dbl> <chr>
1 SUPER_… 8.43e7 8.44e7 ENST… 1000 - 8.43e7 8.44e7 0,0,… 21 96,153…
2 SUPER_… 3.71e7 3.71e7 ENST… 1000 - 3.71e7 3.71e7 0,0,… 5 38,169…
3 SUPER_… 8.19e6 8.19e6 ENST… 1000 + 8.19e6 8.19e6 255,… 1 16,
4 SUPER_… 1.12e8 1.12e8 ENST… 1000 + 1.12e8 1.12e8 255,… 1 55,
5 SUPER_… 8.05e7 8.05e7 ENST… 1000 + 8.05e7 8.05e7 130,… 1 18,
6 SUPER_… 8.87e7 8.87e7 ENST… 1000 - 8.87e7 8.87e7 255,… 2 26,76,
7 SUPER_… 4.65e7 4.65e7 ENST… 1000 + 4.65e7 4.65e7 0,0,… 4 118,94…
8 SUPER_… 1.99e8 1.99e8 ENST… 1000 + 1.99e8 1.99e8 255,… 1 8,
9 SUPER_… 4.11e7 4.11e7 ENST… 1000 + 4.11e7 4.11e7 0,0,… 6 920,18…
10 SUPER_… 1.12e8 1.12e8 ENST… 1000 - 1.12e8 1.12e8 255,… 2 23,109,
# … with 189,508 more rows, 1 more variable: blockLength <chr>, and abbreviated
# variable names ¹thickStart, ²thickEnd, ³blockStartgenome.luc <- read_tsv("../../analyses/makeHub/data/genomes/mMyoLuc1.genome", col_names = c("chr", "length"))Rows: 260 Columns: 2
── Column specification ────────────────────────────────────────────────────────
Delimiter: "\t"
chr (1): chr
dbl (1): length
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.genome.luc# A tibble: 260 × 2
chr length
<chr> <dbl>
1 SUPER__1 244374582
2 SUPER__2 212563729
3 SUPER__3 211792393
4 SUPER__4 121730464
5 SUPER__5 113776203
6 SUPER__6 111457381
7 SUPER__7 98951147
8 SUPER__8 93510542
9 SUPER__9 91912741
10 SUPER__10 84231690
# … with 250 more rowstoga.luc.summary <- toga.luc %>%
group_by(chr) %>%
summarize(n_genes = n_distinct(name)) %>%
left_join(genome.luc, by="chr") %>%
mutate(chr = factor(chr, levels=str_sort(chr, numeric = T) %>% unique))toga.luc.summary %>%
ggplot(
aes(x=length, y=n_genes, label=chr)
) +
geom_text(aes(color=(as.numeric(chr)<=23))) +
theme_pubclean() +
scale_x_log10() +
scale_y_log10() +
ggtitle("TOGA: mMyoLuc1 (hg38)")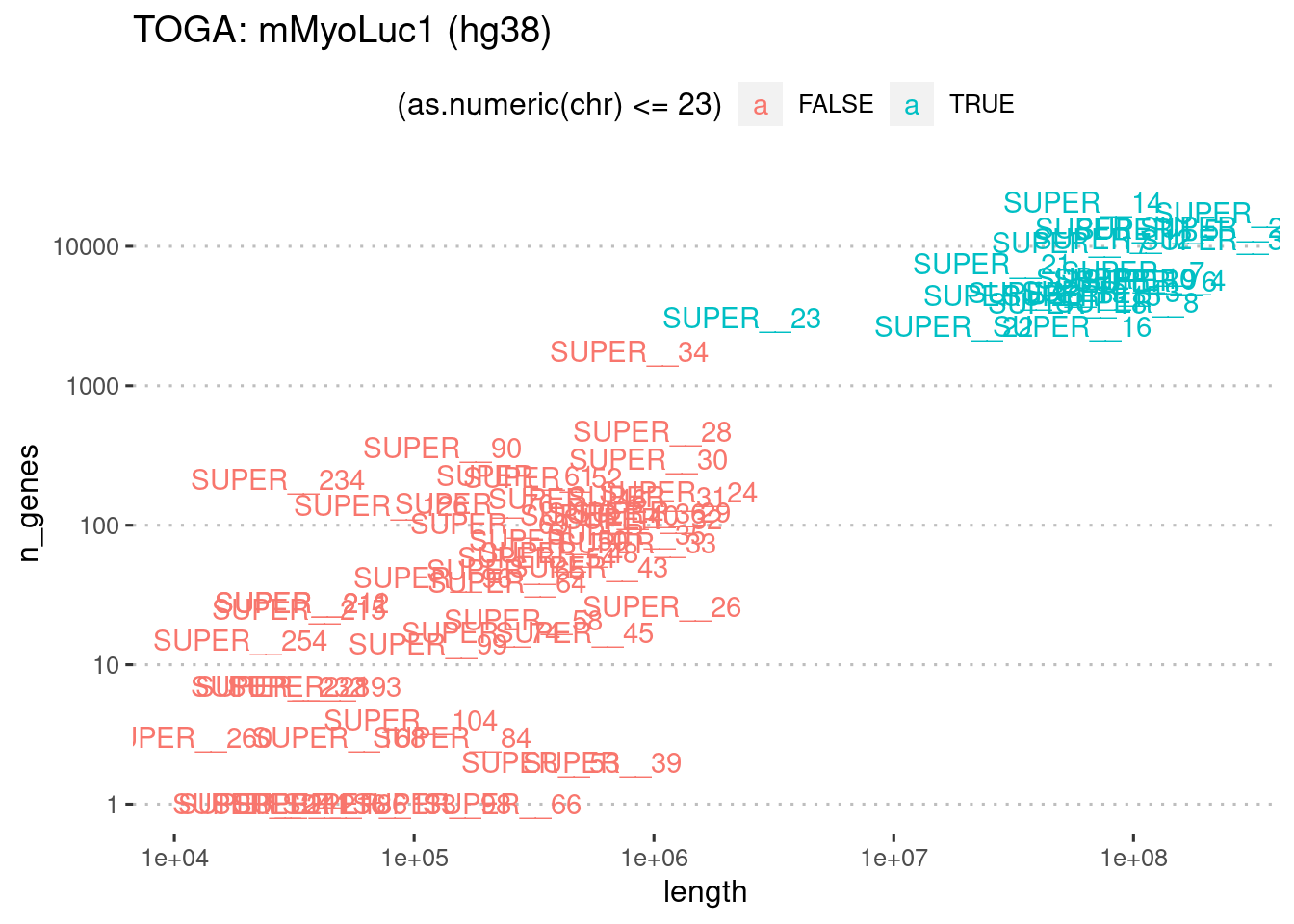
sessionInfo()R version 4.2.2 Patched (2022-11-10 r83330)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.5 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggpubr_0.4.0 naturalsort_0.1.3 forcats_0.5.2 stringr_1.4.1
[5] dplyr_1.0.10 purrr_0.3.5 readr_2.1.3 tidyr_1.2.1
[9] tibble_3.1.8 ggplot2_3.4.0 tidyverse_1.3.2
loaded via a namespace (and not attached):
[1] httr_1.4.4 sass_0.4.2 bit64_4.0.5
[4] vroom_1.6.0 jsonlite_1.8.3 carData_3.0-5
[7] modelr_0.1.9 bslib_0.4.1 assertthat_0.2.1
[10] highr_0.9 googlesheets4_1.0.1 cellranger_1.1.0
[13] yaml_2.3.6 pillar_1.8.1 backports_1.4.1
[16] glue_1.6.2 digest_0.6.30 promises_1.2.0.1
[19] ggsignif_0.6.4 rvest_1.0.3 colorspace_2.0-3
[22] htmltools_0.5.4 httpuv_1.6.6 pkgconfig_2.0.3
[25] broom_1.0.1 haven_2.5.1 scales_1.2.1
[28] later_1.3.0 tzdb_0.3.0 timechange_0.1.1
[31] git2r_0.30.1 googledrive_2.0.0 farver_2.1.1
[34] generics_0.1.3 car_3.1-1 ellipsis_0.3.2
[37] cachem_1.0.6 withr_2.5.0 cli_3.4.1
[40] magrittr_2.0.3 crayon_1.5.2 readxl_1.4.1
[43] evaluate_0.18 fs_1.5.2 fansi_1.0.3
[46] rstatix_0.7.0 xml2_1.3.3 tools_4.2.2
[49] hms_1.1.2 gargle_1.2.1 lifecycle_1.0.3
[52] munsell_0.5.0 reprex_2.0.2 compiler_4.2.2
[55] jquerylib_0.1.4 rlang_1.0.6 grid_4.2.2
[58] rstudioapi_0.14 rmarkdown_2.17 gtable_0.3.1
[61] abind_1.4-5 DBI_1.1.3 R6_2.5.1
[64] lubridate_1.9.0 knitr_1.40 bit_4.0.4
[67] fastmap_1.1.0 utf8_1.2.2 workflowr_1.7.0
[70] rprojroot_2.0.3 stringi_1.7.8 parallel_4.2.2
[73] Rcpp_1.0.9 vctrs_0.5.0 dbplyr_2.2.1
[76] tidyselect_1.2.0 xfun_0.34